

 Previous page
Previous page
Dynamic solid-state properties and phase transitions in organosilicon compounds (X. Helluy, J. Kümmerlen and A. Sebald, in collaboration with W.A. Dollase/Los Angeles and R. Dinnebier/Bayreuth)
Many organosilicon compounds are non-polar, nearly spherically shaped
molecular solids, and display concomitant dynamic solid-state properties.
The molecular compounds E(SiMe3)4, with E = C,Si
belong to this category and exist as crystalline solids at ambient temperatures.
Previously, only the gas phase structures had been determined for these
two compounds and both have molecular T symmetry. For E = Si the crystal
structure has now been determined to have space group Fm3m by Rietveld
refinement of powder X-ray diffraction data obtained at room temperature.
Both compounds E(SiMe3)4 undergo structural phase
transitions in the temperature range 200 to 220 K. In the respective low-temperature
phases the molecular dynamic properties of E(SiMe3)4 were
characterized by variable-temperature 13C and 29Si
one- and two-dimensional CP/MAS NMR. Quantitative evaluation of the respective
exchange rate constants for molecular reorientation and for internal reorientation
of the SiMe3 groups as a function of temperature reveals striking
differences in the dynamic properties of these two chemically closely related
methane homologues. For Si(SiMe3)4 at low temperatures,
internal reorientation of the SiMe3 groups is observed, and
at higher temperatures additional reorientation of the entire molecule
around its preferred molecular axes takes place. For C(SiMe3)4,
the thermally activated modes of reorientation in the solid state are quite
different: over the temperature range of the low-temperature phase, no
internal reorientation of the SiMe3 groups takes place, and
only reorientation of the entire molecule is observed. A possible explanation
for this difference in thermally activated molecular dynamics is offered
by force field calculations, based on the gas-phase structures of the two
molecules: due to higher intramolecular steric hindrance in C(SiMe3)4,
whole-molecule reorientation may be the preferred reorientational mode
for this compound (Fig. 3.8-10).
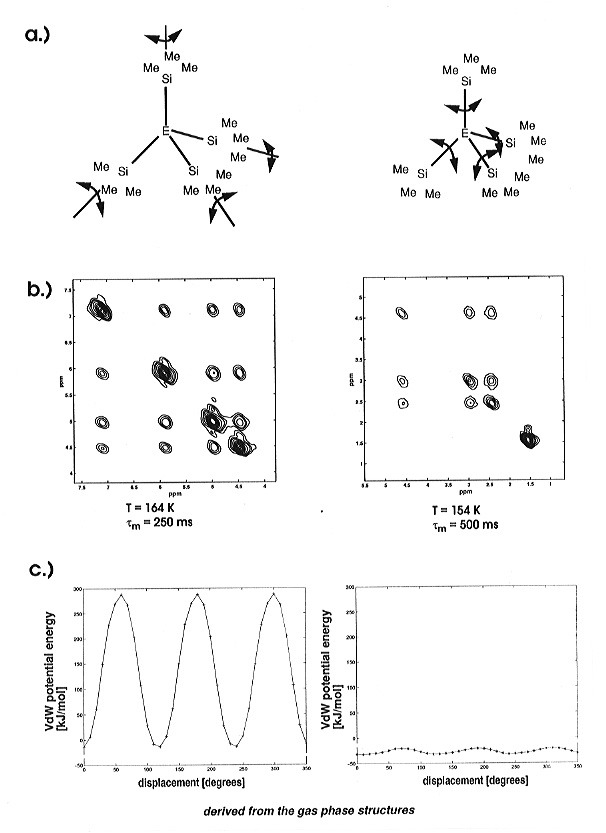 |
|
a) schematic illustration of reorientational modes in solid E(SiMe3)4 b) 13C 2D exchange spectra for E = C (left) and E = Si (right) c) force-field calculations of activation barriers for internal SiMe3 reorientation for E = C (left) and E = Si (right). |
Numerically exact spectral lineshape simulations for isolated 2-
and 3-spin systems under MAS conditions (S. Dusold and A. Sebald)
Isolated 2- and 3-spin systems occur (either naturally, or man-made
by isotopic labelling) in many different chemical compounds, ranging from
biochemicals to inorganic solids. One class of chemicals where such spin
systems naturally occur are transition-metal phosphine complexes (the M-31P
and M(31P)2 fragments, where M represents a spin-1/2
isotope such as 113Cd, 195Pt, or 199Hg).
The Hamiltonian appropriate to describe isolated dipolar coupled 2- and
3-spin systems under MAS conditions does not commute with itself at different
times. The practical consequences in MAS NMR spectra of such spin systems
are observed as complicated spectral lineshapes, strongly dependent on
the external magnetic field strength and applied MAS frequency. Interpretation
of such MAS NMR spectra "by inspection" is not possible. Their spectral
lineshapes not only depend on the magnitudes of NMR interactions but also
depend on the mutual orientations of the various interaction tensors. Hence,
numerically exact spectral lineshape simulations of MAS NMR spectra of
polycrystalline powder samples can be used to yield information about the
orientation of e.g. chemical shielding tensors in the principal axes system
of a molecule or crystal, or in the determination of internuclear distances.
Conventional MAS NMR of polycrystalline powder samples, in conjunction
with numerically exact spectral lineshape simulations thus provide information
which would otherwise require NMR experiments on oriented single crystals.
A complete analysis of the MAS NMR spectra of isolated dipolar coupled
2- and 3-spin systems lifts a severe limitation with regard to sample availability
for solid-state NMR, as no single crystals are necessary. With our focus
on optimizing efficiency, we have developed computational methods that
allow for high-quality iterative fitting of the experimentally observed
MAS NMR spectral lineshapes. We illustrate this technique with the example
of double- and triple-resonance experimental and calculated 113Cd
CP/MAS NMR spectra of a representative cadmium-phosphine complex (Fig.
3.8-11).
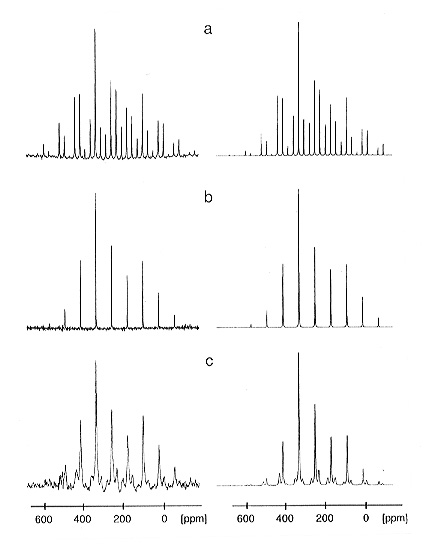 |
|
a) conventional 113Cd CP/MAS NMR spectrum; b) 113Cd CP/MAS NMR spectrum with additional high-power 31P decoupling; c) 113Cd CP/MAS NMR spectrum with additional low-power 31P recoupling. The most shielded component of the 31P chemical shielding tensors is typically oriented close to the P-Cd bond direction in the molecular frame. |
Unambiguous confirmation of space group Pa3 for the pseudo-cubic phase of SiP2O7 by means of 31P NMR (J. Kümmerlen and A. Sebald, in collaboration with R. Iuliucci and B.H. Meier/Nijmegen)
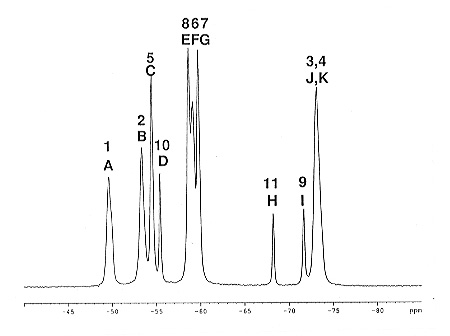
The single-crystal X-ray structure of the pseudo-cubic phase of SiP2O7 has been refined by simulation and space group Pa3 has been proposed as the correct solution of the structure determination in the literature. This space group requires the presence of eleven crystallographically inequivalent P sites in the asymmetric unit, and for reasons of symmetry, two of the P2O7 units must contain linear P-O-P moieties. The validity of the proposed structure solution has been questioned because of the required presence of supposedly unfavorable linear P-O-P arrangements (amongst other, non-linear P-O-P arrangements). In order to resolve this remaining ambiguity concerning the correct space group of the pseudo-cubic phase of SiP2O7, 31P MAS NMR experiments which only report distance connectivities but are widely insensitive to chemical shifts, must be carried out. Such solid-state rf-driven spin diffusion 31P NMR experiments have been carried out, and the distance connectivities (short, intermediate, long) as seen by 31P NMR are compared to the connectivities as required by the proposed space group from single-crystal X-ray diffraction. There are 11! = 39916800 assignment permutations for the various 31P resonances to the eleven inequivalent P sites in the asymmetric unit. By using formalised "short, intermediate, and long" distance constraints from the 31P solid-state NMR experiments, space group Pa3 can be confirmed by NMR, including an unambiguous assignment of the 31P resonances to the respective P sites in the asymmetric unit. Figure 3.8-12 shows a 31P MAS NMR spectrum of SiP2O7, and the resulting model-free assignment of 31P resonances (A-K) to the crystallographic P sites (1-11).
Determination of anisotropy of long-range J-coupling by 119Sn
OMAS NMR (C. Marichal and A. Sebald)
Isotropic J coupling, an interaction transferred through electrons
in chemical bonds, is widely exploited in liquid state NMR for purposes
of structure elucidation. Like all other NMR interactions, however, J
coupling is an anisotropic interaction, described by a second-rank tensor,
such that in nonviscous solutions it is motionally averaged to its isotropic
value. In principle, anisotropy of J in crystalline solids should
be observable. However, J coupling interactions in solids are difficult
to observe experimentally as they are usually masked by the simultaneous
presence of multiple dipolar couplings much larger in magnitude. So far,
only few attempts to determine the anisotropy of J coupling, mostly
by means of single-crystal NMR, have been reported in the literature. All
of these experimental studies have considered J coupling over a
single bond, and to the best of our knowledge no experimental data on the
anisotropy of long-range J coupling over more than one bond have
been reported. We have
 |
|
|
investigated the anisotropy of 2J(119Sn,
117Sn) in solid (benzyl3Sn)2O, using 119Sn
off-magic-angle spinning (OMAS) NMR techniques in conjunction with spectral
lineshape simulations. Our finding of considerable anisotropy of 2J(119Sn,
117Sn) bears relevance on various other solid-state NMR techniques,
designed to determine internuclear distances from NMR experiments: if J
anisotropy is present but not taken into account in the analysis of such
NMR experiments, the internuclear distances derived will be inaccurate.
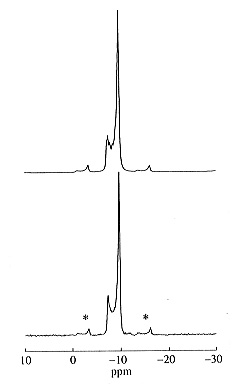
Figure 3.8-13 shows a comparison of the experimental and the calculated
119Sn OMAS NMR spectra from which we derive an anisotropy of
2J(119Sn, 117Sn) = 850 ±
300 Hz, similar to the magnitude of |
2Jiso(119Sn, 117Sn)|
= 955 Hz.
 |
|
|

Tel: +49-(0) 921 55 3700 / 3766, Fax: +49-(0) 921 55 3769, E-mail: bayerisches.geoinstitut(at)uni-bayreuth.de